Single-cell
How to Choose Leiden Resolution: Comparing leiden, auto_resolution, and champ
When running single-cell clustering, almost everyone eventually asks the same question: what should the Leiden resolution be?
If the value is too low, distinct biological populations can be merged. If it is too high, a stable cell population can be fragmented into many small clusters. The numbers themselves, such as 0.5, 1.0, or 1.2, do not have intrinsic biological meaning; they are controls for the granularity of community detection.
This tutorial asks a practical question: which OmicVerse tools can help choose a Leiden resolution?
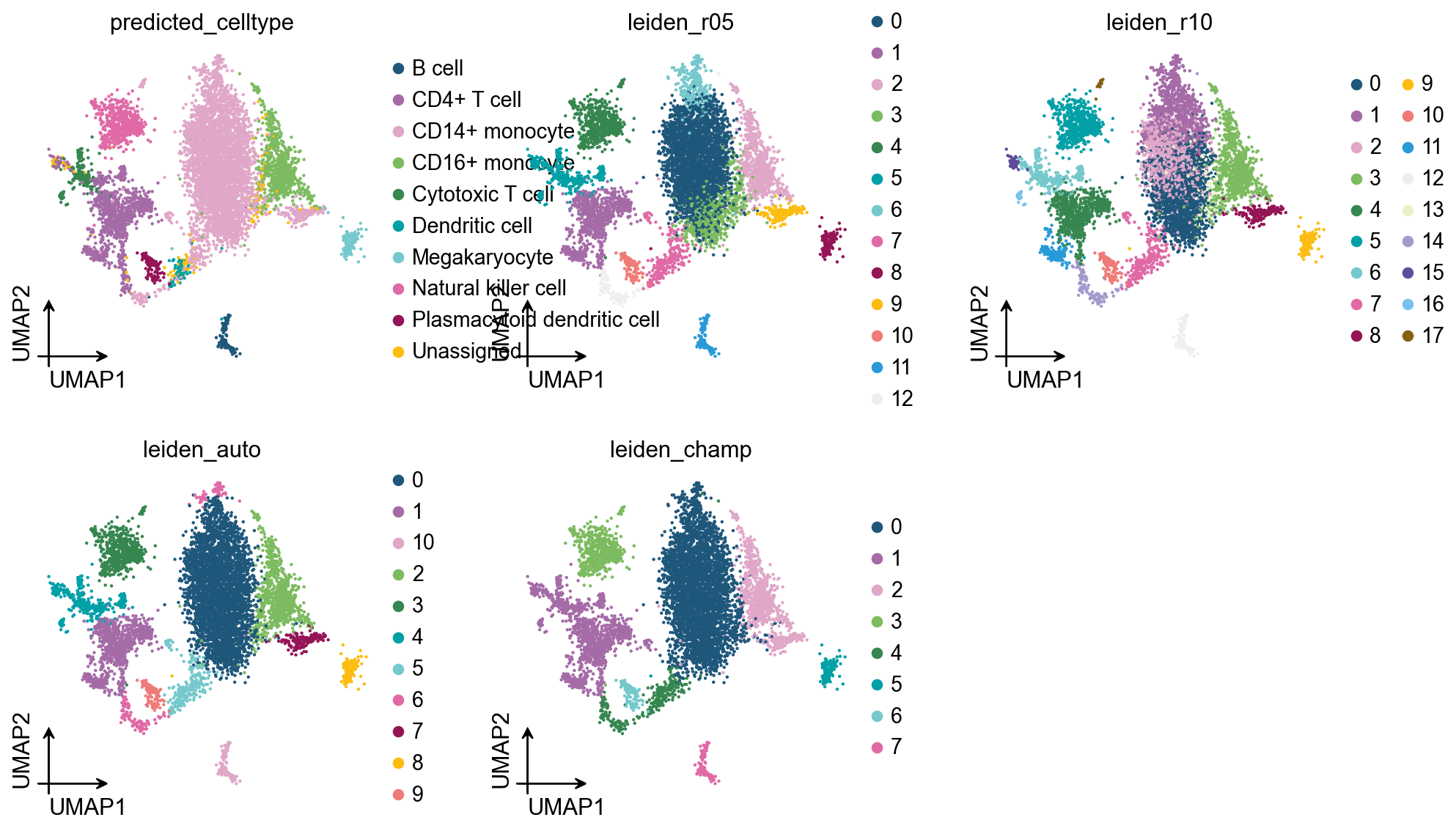
We use ov.datasets.pbmc8k() because it includes curated predicted_celltype labels, making it possible to compare clustering results against a biological reference.
| Method | Function | Main idea |
|---|---|---|
| Manual Leiden | ov.pp.leiden(adata, resolution=r) | The user chooses the resolution directly |
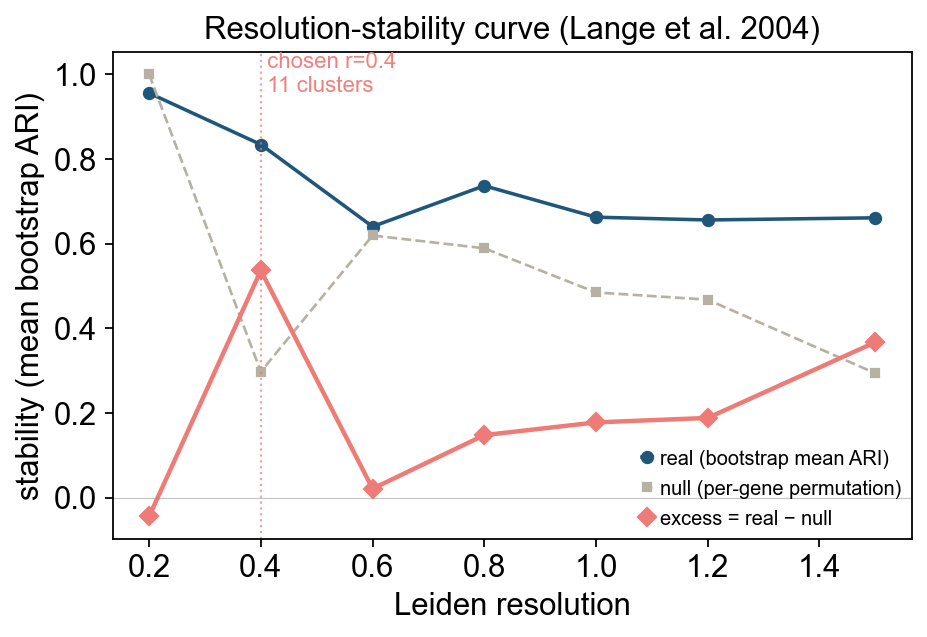
| bootstrap-ARI | ov.single.auto_resolution() | Choose the resolution that is most stable under data perturbation |
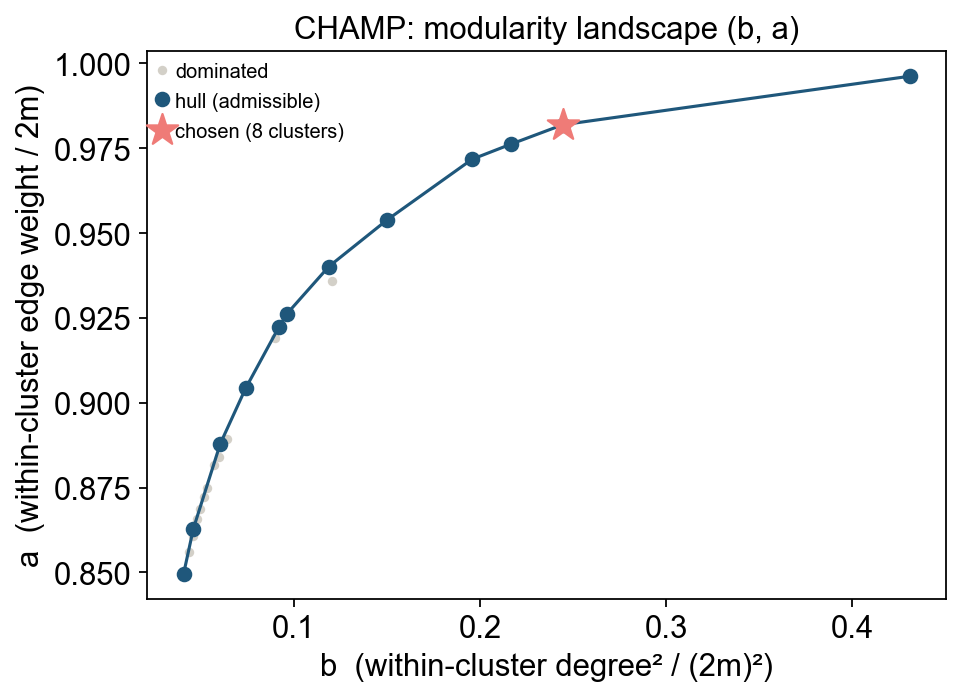
| CHAMP | ov.single.auto_resolution(method='champ') | Choose the partition that is most stable across the modularity landscape |
The key point is that auto_resolution and champ answer different questions. The bootstrap-ARI workflow asks which resolution is stable under resampling. CHAMP asks which partition is stable as the gamma parameter changes. Neither method replaces biological interpretation; both provide stronger starting points than an arbitrary default.
Environment setup
import os, io, contextlib
os.environ.setdefault("OMICVERSE_DISABLE_LLM", "1")
import warnings
warnings.filterwarnings("ignore")
import numpy as np
import pandas as pd
import matplotlib.pyplot as plt
from sklearn.metrics import adjusted_rand_score, normalized_mutual_info_score
import omicverse as ov
ov.style()Load and preprocess PBMC8k
adata = ov.datasets.pbmc8k()
adata.var_names_make_unique()The dataset includes predicted_celltype, so later results can be scored against the curated cell type annotation.
adata = ov.pp.qc(
adata,
tresh={"mito_perc": 0.2, "nUMIs": 500, "detected_genes": 250},
doublets_method="scrublet",
)
ov.pp.preprocess(adata, mode="shiftlog|pearson", n_HVGs=2000)
adata.raw = adata
adata = adata[:, adata.var.highly_variable]
ov.pp.scale(adata)
ov.pp.pca(adata, layer="scaled", n_pcs=50)
ov.pp.neighbors(adata, n_neighbors=15, use_rep="scaled|original|X_pca")
ov.pp.umap(adata)After QC, the example retains 7,677 cells and 2,000 highly variable genes. All methods use the same neighbor graph, so the comparison is controlled.
Baseline: manual Leiden
ov.pp.leiden(adata, resolution=0.5, key_added="leiden_r05")
ov.pp.leiden(adata, resolution=1.0, key_added="leiden_r10")In this run, resolution=0.5 produced 13 clusters and resolution=1.0 produced 18 clusters. That behavior is expected: higher resolution usually means more clusters. The problem is that more clusters do not necessarily mean better biology.
Bootstrap-ARI with auto_resolution
ov.single.auto_resolution() uses null-adjusted bootstrap-ARI stability by default. It repeatedly subsamples and reclusters at candidate resolutions, then subtracts a null background estimated from permuted data.
with contextlib.redirect_stdout(io.StringIO()):
_, autores_best, autores_scores = ov.single.auto_resolution(
adata,
resolutions=[0.2, 0.4, 0.6, 0.8, 1.0, 1.2, 1.5],
n_subsamples=5,
n_null_subsamples=3,
random_state=0,
key_added="leiden_auto",
)
print(f"auto_resolution chose r = {autores_best}")In the PBMC8k example, auto_resolution selected r = 0.4.
CHAMP
CHAMP follows a different logic. Instead of asking which resolution survives bootstrap perturbation, it looks for stable partitions across the modularity landscape.
_, champ_best_r, champ_df = ov.single.auto_resolution(
adata,
method="champ",
key_added="leiden_champ",
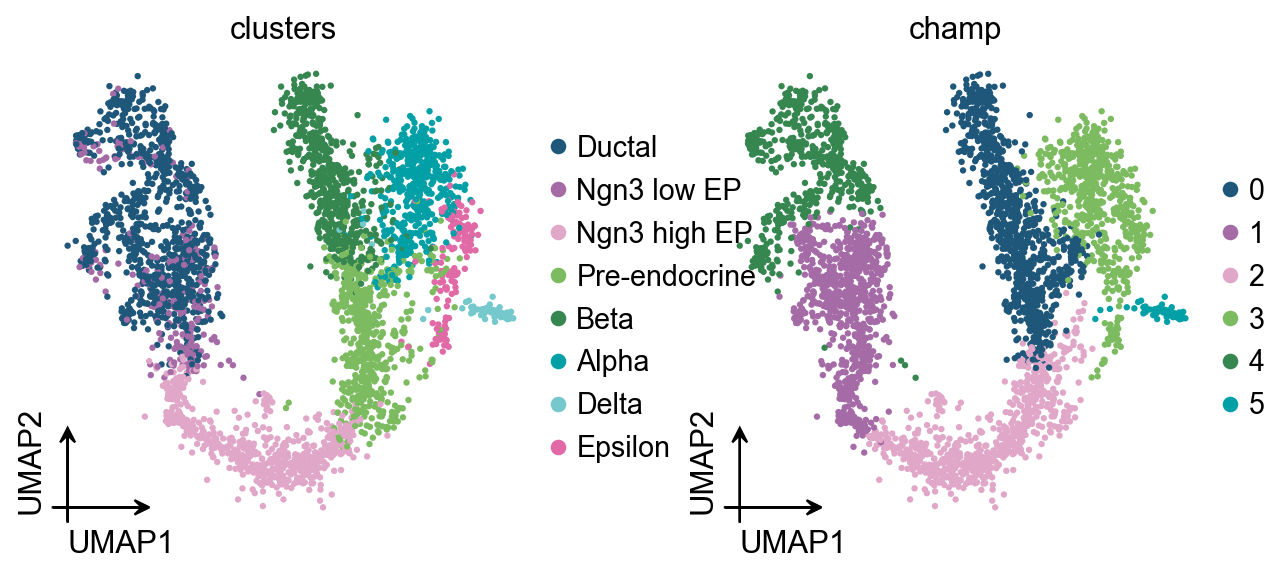
)In the same PBMC8k run, CHAMP returned an equivalent Leiden resolution around r = 0.356, with a stable gamma range near [0.08, 0.21].
What the comparison showed
On PBMC8k, the automatic methods performed better than manually choosing resolution=0.5 or resolution=1.0. CHAMP had a slightly higher ARI, while bootstrap auto_resolution had a slightly higher NMI.
| Method | Clusters | ARI | NMI |
|---|---|---|---|
| Manual Leiden, r=0.5 | 13 | lower | lower |
| Manual Leiden, r=1.0 | 18 | lower | lower |
| auto_resolution, r=0.4 | 11 | 0.822 | 0.807 |
| CHAMP, gamma in [0.08, 0.21] | 8 | 0.829 | 0.794 |
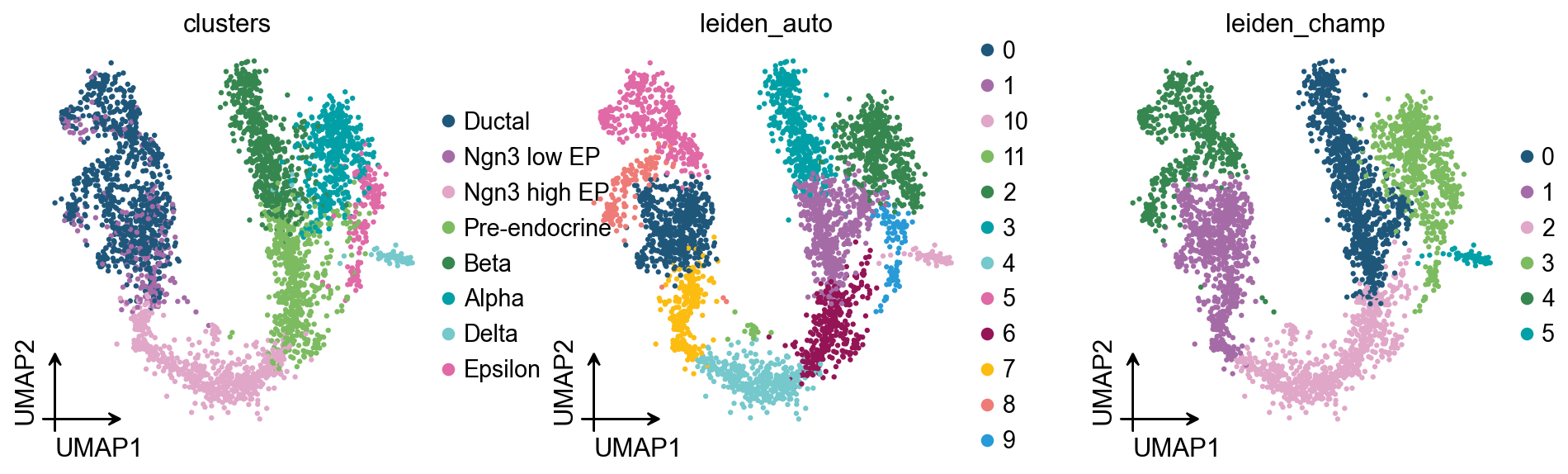
The practical reading is simple: automatic resolution selection is useful, but it should be treated as a reproducible starting point. Marker genes, known cell states, sample design, and downstream biological questions still decide whether a clustering is useful.
Figures