Open Ecosystem Initiative | OmicVerse 2.1 Release Update
Editor's note
Starting an open source community in China is difficult. Because of complex interests and incentives, it is hard for us to directly copy ecosystems such as scverse or Bioconductor. For a long time, the only OmicVerse developers were myself, Leihu, and Xuehai.
In many cases, we would read about an interesting algorithm, then run into installation constraints. So we would remove strict version limits, migrate the package into the external module, and call it directly through OmicVerse. Sometimes we would also improve the package's output visualization, for example by adding progress bars or better colors. If a package was easy to install, we would introduce its algorithm function into existing OmicVerse common interfaces such as
ov.single.Annoandov.single.Traj, so users could easily keep using OmicVerse downstream analysis and visualization functions.The arrival of the AI era has sent us a different signal: it now seems that everyone can become a developer. If you tell AI what you need, you can rewrite an original R package in a different language. Previously, even rewriting DESeq2-style functionality could become a Bioinformatics paper. Therefore, as the main maintainer of OmicVerse, I would like to formally invite everyone to join this open ecosystem.
At the same time, I have written articles such as "the best single-cell tutorial." As another part of the open source community, we will write analysis tutorials based on the OmicVerse ecosystem, with the goal of lowering the entry barrier for beginners as much as possible.
The OmicVerse v2.1 update is also very interesting. It adds py-monocle2, microbiome analysis, metabolomics analysis, and many other features. Details are listed below.
steorra
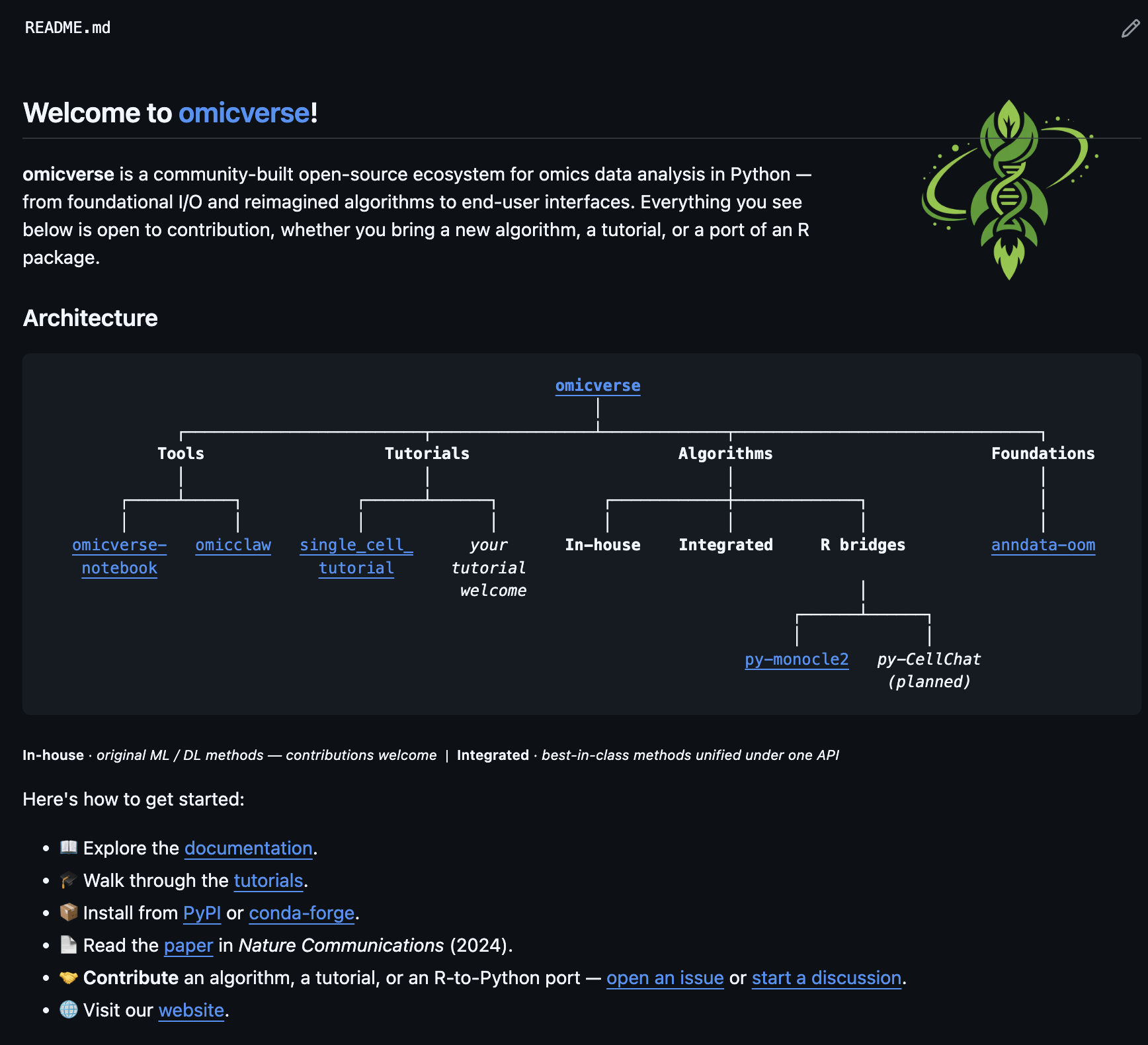
Project homepage: https://github.com/omicverse
For Developers
Because I am still active in frontline development, I can promise that after an algorithm refactoring is completed, we can discuss how to publish it in a top journal together. Friends who know me well know that I do not compete for first authorship. Related OmicVerse series papers, such as Nature Protocols-style articles, will also include names according to author contributions.
As an economic promise, if our open source community receives donations, we will use that money to give back to all developers according to contribution, although I think the probability of receiving donations is extremely low.
Contribution Guide
If you are interested, you can add my contact FernandoZeng. I will invite you into the developer group and send a GitHub Member invitation.
For repositories built by authors, you will have full administrator permissions. In other words, you can transfer repositories you created back to your personal account at any time. I think depriving ownership is not a good practice for an open source community.
After the repository is built, we can discuss how to connect it into OmicVerse common functions, making it easier for bioinformatics researchers and developers to call and use.
Refactoring Guide
If you rewrite an analysis package in Python or Rust, you can create a repository under the organization named like rust-monocle2 or py-monocle2. For the README and package format, you can refer to what we have already built:
py-monocle2: https://github.com/omicverse/py-monocle2py-DoubletFinder: https://github.com/omicverse/py-DoubletFinder
For Ecosystem Users
I know not everyone wants to write code, but I believe many people still want to participate in building an open source community or ecosystem. A single spark can start a prairie fire.
At the end of this article, I will provide a user group. If there is an algorithm you want to add to OmicVerse, please do not hesitate to tell us in time. In the current new version, we added microbiome and metabolomics analysis, and proteomics will come later. At the algorithm level, we added visualization improvements for CellPhoneDB, a Python refactoring of monocle2, GPU refactoring for algorithms such as pca, and more. These were all requests raised by users, and we implemented them one by one in the new version.
You can also submit a feature request issue in the OmicVerse repository. Over the past three years, we have added more than 100 features from issues.
v2.1 Release Update
If you only look at the version number, many people may think this is just a normal minor release. But if you read the English release notes, you will see that this is actually a major iteration. From v2.0.0 to the current development tree, master has accumulated 462 commits, 429 changed files, and nearly 100,000 lines of new code. This does not include two new branches still in the merge window: one for metabolomics and one for 16S / microbiome.
In this update, we mainly added and optimized single-cell trajectory inference, spatial omics, preprocessing and I/O, the plotting system, the Agent runtime, and new multi-omics modules.
We also added the ov.metabol metabolomics module, added ov.micro and ov.alignment for 16S / microbiome analysis, and completed high-resolution spatial omics workflows for Xenium, Visium HD, and CosMx.
This is a major version update. Please update with:
pip install -U omicverse1. Update Overview
PP Module
- Added
pydoubletfinderandscdblfinderbackends for doublet detection without requiring an R environment. - Automatically detects mitochondrial prefixes, so users no longer need to manually distinguish
MT-andmt-. - Upgraded Harmony to
harmonypy v0.2.0, supporting GPU, CPU NumPy, and Apple Silicon MLX backends. - Optimized memory handling for
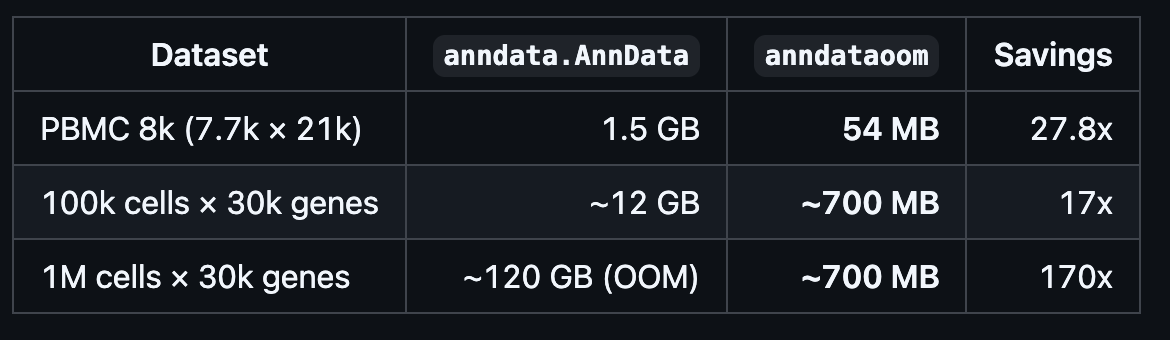
torch_pcaandcovariance_eigh, reducing OOM caused by dense conversions. - Added the Rust OOM backend
anndataoomfor out-of-memory AnnData reading.
Single Module
- Added
ov.single.Monocle, a pure Python refactoring of Monocle 2 withoutrpy2or an R environment. - Further strengthened dynamic trajectory tools, including lineage-aware trend plotting and optimized Palantir / Slingshot visualization.
Space Module
- Added
ov.io.read_xeniumfor complete Xeniumouts/directory loading. - Supports converting Xenium
cell_boundariesto WKT polygons, which can be passed directly toov.pl.spatialseg. - Added
ov.io.write_visium_hd_cellsegfor exporting back to a SpaceRanger v4-compatible directory structure. - Added CosMx FOV-aware plotting with multi-FOV layout and per-FOV overlay.
- Added the CellSAM backend, and officially renamed
stardist()tocellseg(). - Added
method='cellcharter'toov.utils.cluster.
Pl Module
- Added a Marsilea-based heatmap plotting API.
- Added a full set of cell-cell communication plotting APIs.
- Added
ov.pl.create_custom_colormap. - Added half-violin boxplots and gradually replaced old plotting interfaces.
- Optimized legend behavior in subset plotting to avoid unused categories interfering with display.
Utils Module
- Added
method='pymclustR'toov.utils.cluster, completely replacing the previousmclust_R. - Added utility functions such as gene ID conversion.
Agent Module
- Fully refactored the
ov.Agent/ Jarvis / OVAgent runtime. - Added Codex OAuth support.
- Migrated multiple message channels into a unified
MessageRuntime. - Completed lower-level capabilities such as sandboxing, timeout, stdout guard, retry, and security hardening.
Metabol Module
- Added the
ov.metabolmodule. - Supports ID mapping, MSEA, Mummichog, SERRF, DGCA, ASCA / MixedLM, ROC / biomarker panel, and other analysis workflows.
Microbiome Module
- Added
ov.microandov.alignment. - Supports a complete workflow from 16S amplicons to microbiome AnnData analysis.
2. Update Details
2.1 Added ov.single.Monocle, a Pure Python Refactoring of Monocle 2
We added Monocle to ov.single. This is not a thin wrapper around an R interface. Instead, the Monocle 2 trajectory inference workflow has been implemented directly in pure Python. This means you no longer need to install rpy2 or maintain a separate R environment.
This refactoring includes:
- DDRTree
- MST
- branch-state assignment
- BEAM differential expression
We did not stop at "it runs." During implementation, we also addressed several performance bottlenecks in the original Monocle 2. For example, the default method='fast' DDRTree is faster, and _project_cells_to_mst no longer uses an O(N²) dense distance matrix. It now uses Delaunay plus sparse MST.
In benchmark datasets, the current implementation reaches a pseudotime correlation above 0.99 with R Monocle 2, while improving speed by 30 to 100 times.
Repository: https://github.com/omicverse/py-monocle2

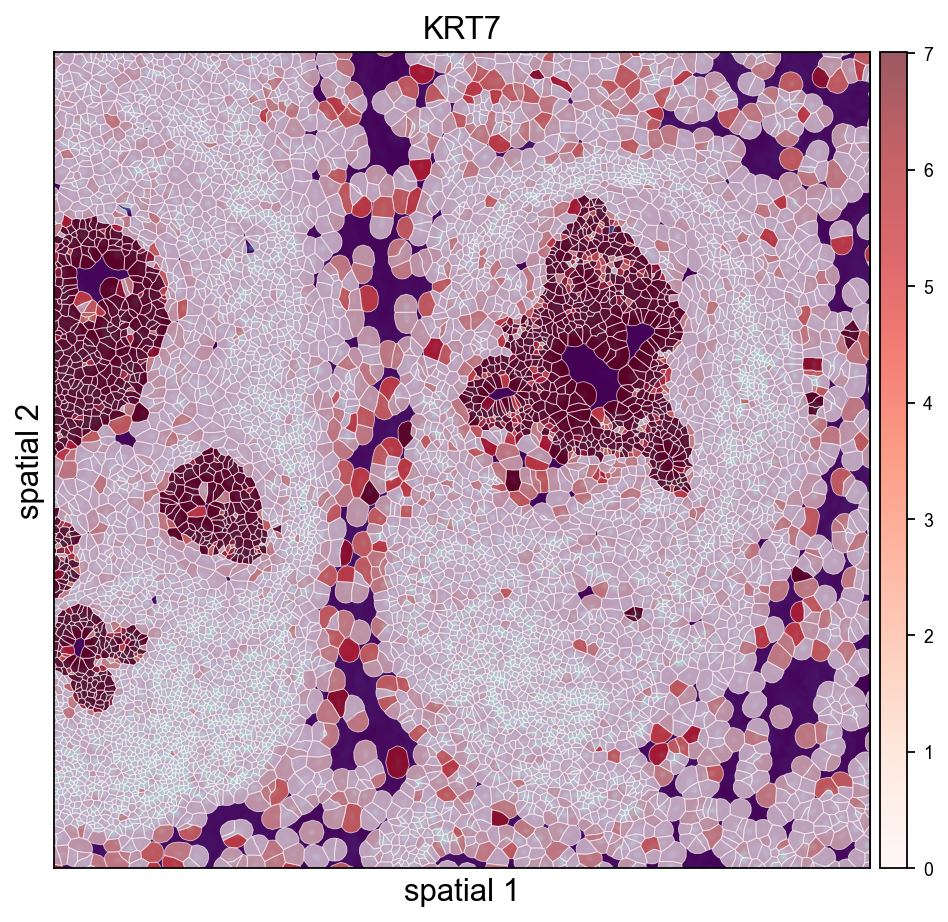
2.2 Completed the Xenium Analysis Workflow
In this release, we officially added ov.io.read_xenium.
In earlier versions, Xenium was already a very common high-resolution spatial platform, but OmicVerse still lacked a truly complete loading and downstream visualization workflow. This gap is now mostly filled.
ov.io.read_xenium currently supports:
cell_feature_matrix.h5cells.csv.gz / cells.parquetexperiment.xeniumcell_boundaries.parquet
The key point is that we convert cell_boundaries into per-cell WKT polygons, which can be connected directly to ov.pl.spatialseg for cell-boundary visualization.
This means Xenium in OmicVerse is no longer merely "a readable format." It can finally run through a complete path:
read -> preprocess -> spatialseg overlayTutorial: Xenium tutorial
2.3 Export Visium HD Back to the SpaceRanger v4 Structure
This is a very practical update, although its value may not be obvious at first glance.
The new version adds ov.io.write_visium_hd_cellseg, which can export cell-level AnnData back into a SpaceRanger v4-compatible directory structure. After completing Cellpose / CellSAM segmentation in OmicVerse, the result no longer stays only inside the analysis object. It can be written back into the 10x ecosystem and continue to connect with Loupe Browser or spaceranger-aware pipelines.
This matters because it moves OmicVerse from "able to analyze data" toward "able to write back into the ecosystem."
We also rewrote the Cellpose tutorial and added a Cellpose vs CellSAM comparison.
Tutorials:
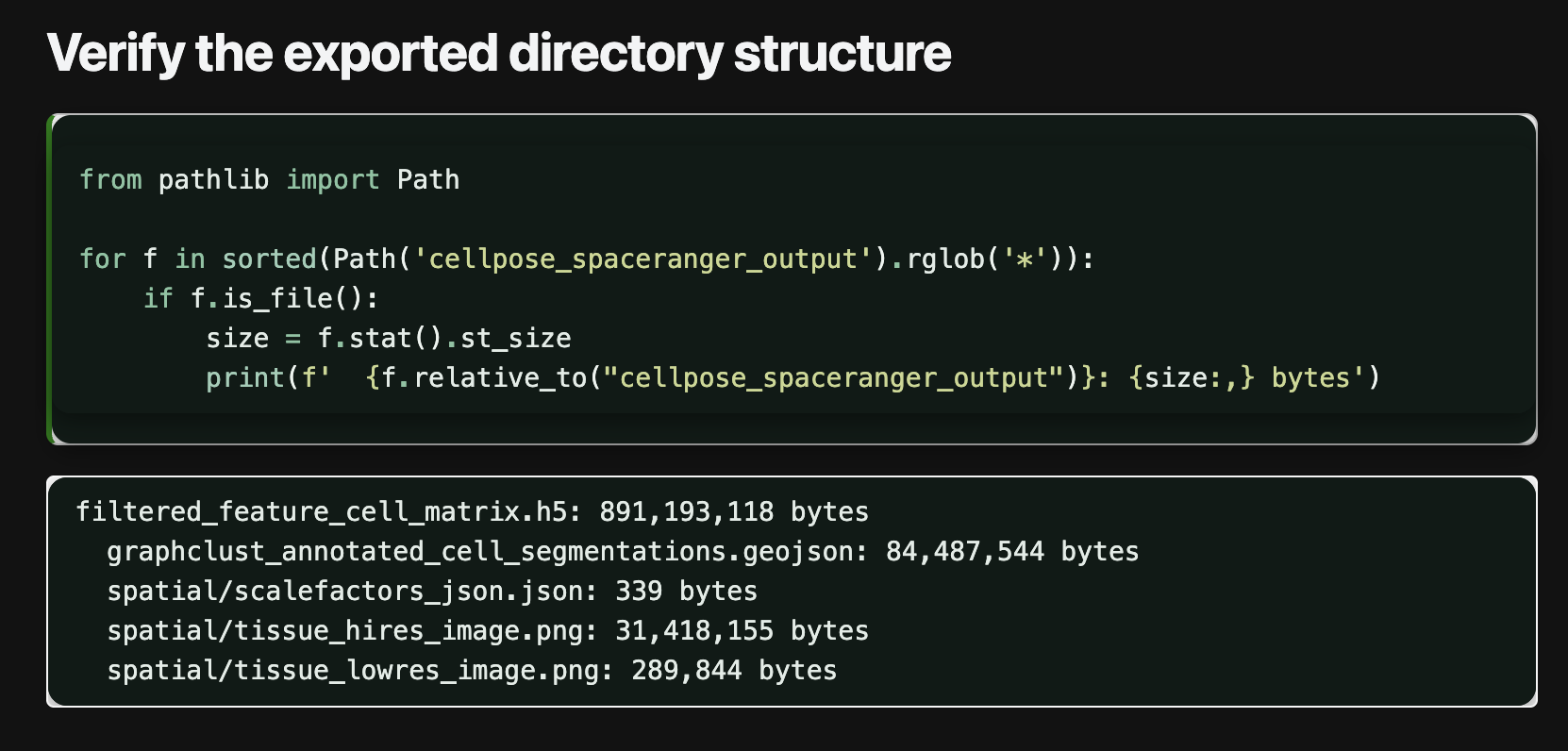
2.4 Completed CosMx FOV-aware Plotting
The CosMx workflow also received many additions.
The new version adds a set of FOV-aware plotting capabilities, including:
- multi-FOV layout
- per-FOV background image overlay
- rasterisation options for dense slides
If you have worked with CosMx, you know these capabilities are not just "a few more parameters." CosMx data is naturally structured as multiple FOVs, large images, and dense cells. Whether plots are easy to read is itself part of the analysis experience.
Tutorial: NanoString / CosMx tutorial
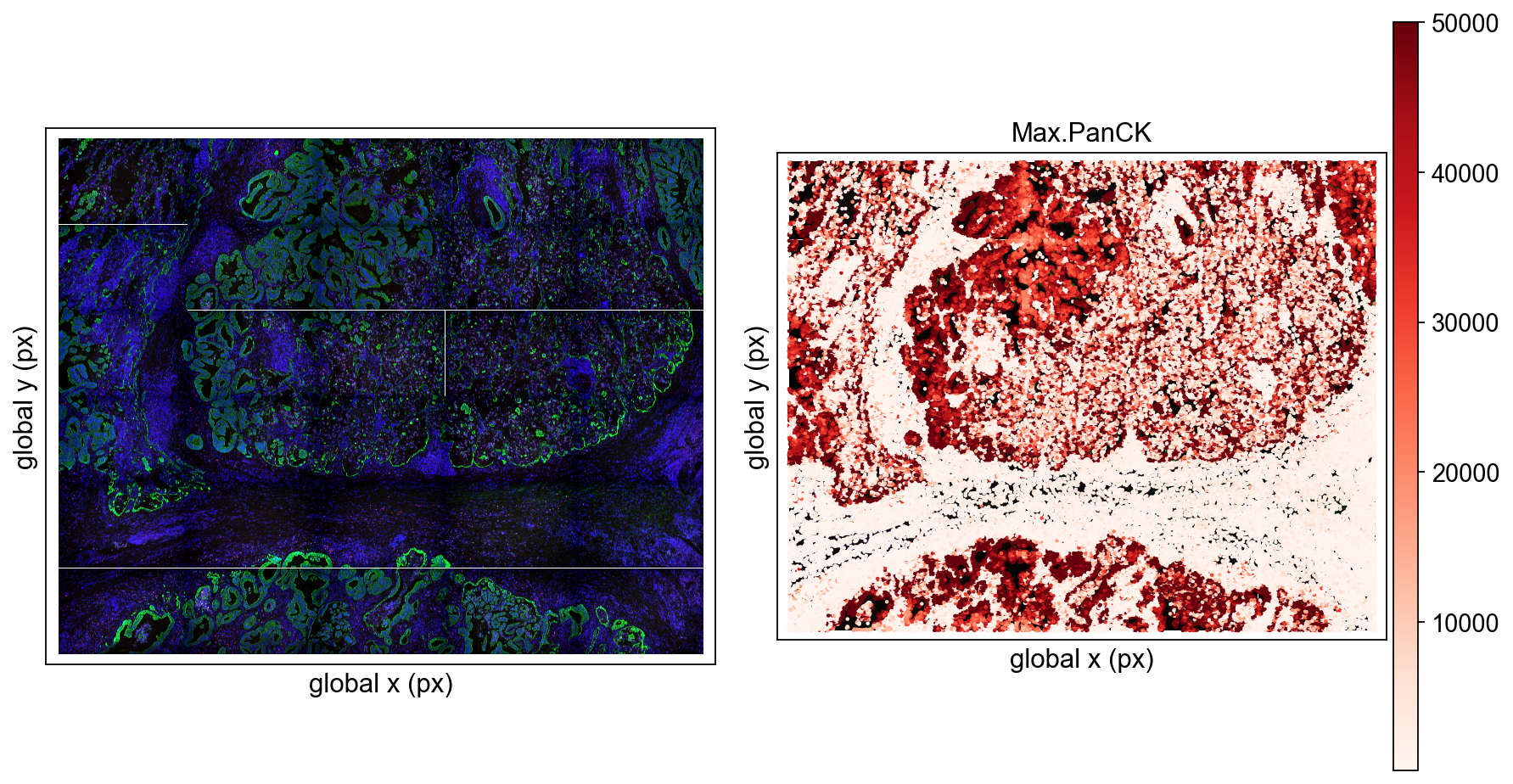
2.5 Added pydoubletfinder and pyscDblFinder to ov.pp.qc, and Continued Improving Preprocessing
In the preprocessing module, we added pydoubletfinder and py-scDblFinder backends.
This means you can now perform doublet detection directly in Python, without configuring a separate R dependency for DoubletFinder.
We also made several small improvements that strongly affect daily use:
- automatic mitochondrial prefix detection;
- optimized memory handling for
torch_pca; - adjusted the default behavior of
scale()to reduce repeated sparse / dense switching; - improved multi-backend support for Harmony;
- added the Rust backend
anndataoomfor out-of-memory AnnData. I will write a separate tutorial about how to useanndataoom.
These may not look like headline features, but in real projects, these lower-level details often determine whether the workflow feels smooth.
Tutorial: CPU / GPU mixed preprocess tutorial
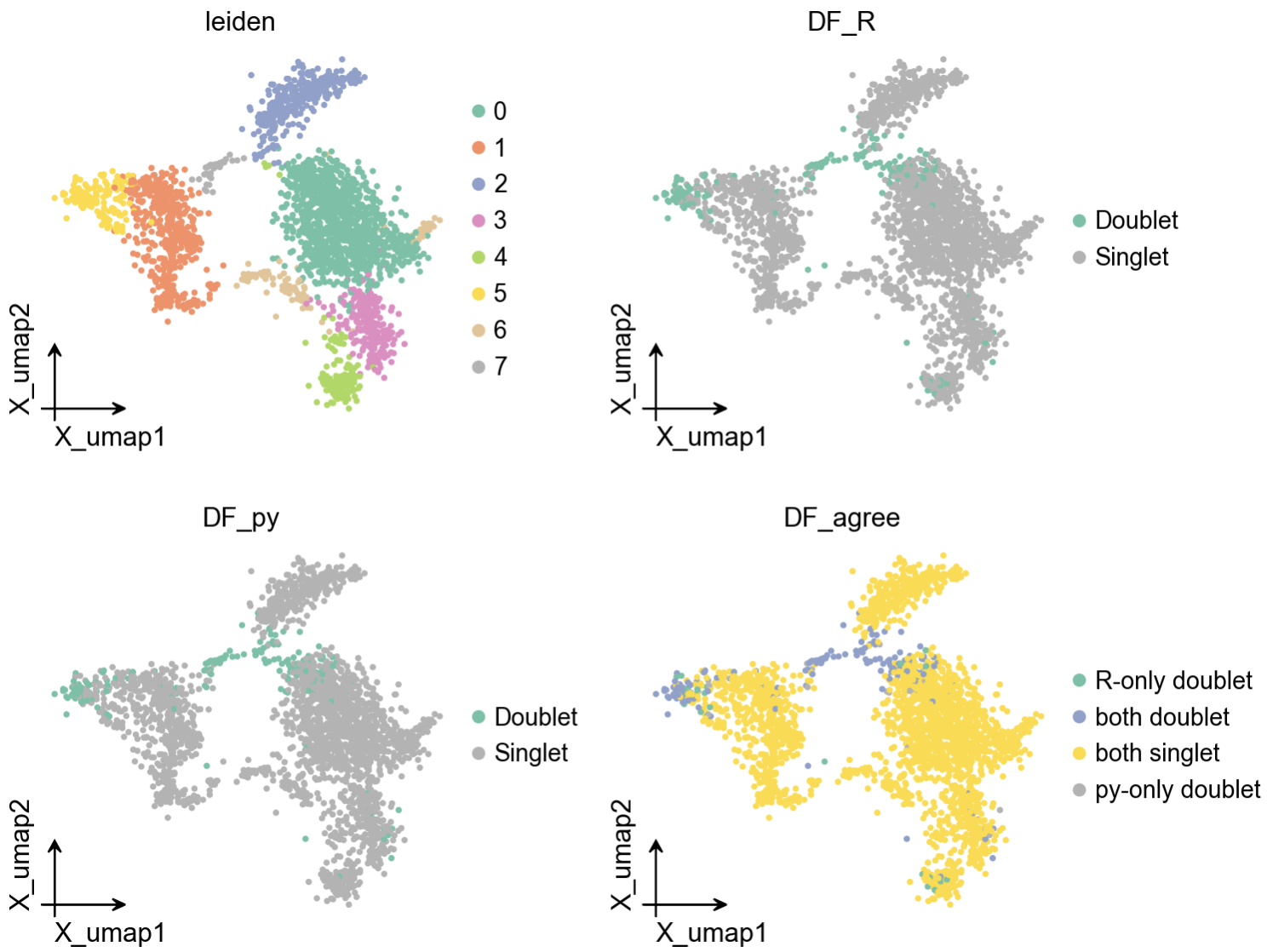
2.6 Plotting System Unification: New Heatmap and CCC Plotting APIs
In this version, ov.pl continues moving toward a unified interface.
Special thanks to Mengxu, the author of scop. With his help, OmicVerse cell-cell communication visualization has been greatly optimized. We will publish a dedicated article later to explain this part of the update:
- added a Marsilea-based heatmap plotting API;
- added a full set of cell-cell communication plotting APIs;
- added
ov.pl.create_custom_colormap; - added half-violin boxplots;
- optimized subset plotting and legend logic;
- organized lazy loading and optional dependency handling for plotting imports.
In short, these updates are not about "adding a few more plots." They are about making OmicVerse plotting feel like a unified system, rather than a pile of historical functions stacked together.
Tutorial: CellPhoneDB plotting tutorial


2.7 Major Improvements to Trajectory Analysis Visualization
We all know that for pseudotime analysis, appropriate visualization can convey much more information. In previous versions, after ov.single.Traj calculated pseudotime, there was only one additional PAGA plot to visualize cell-state transitions.
In version 2.1.2, with Mengxu's help, OmicVerse added many visualization modules similar to scop. We welcome everyone to try them in the new version, and we will later publish a more detailed tutorial on how to use them.


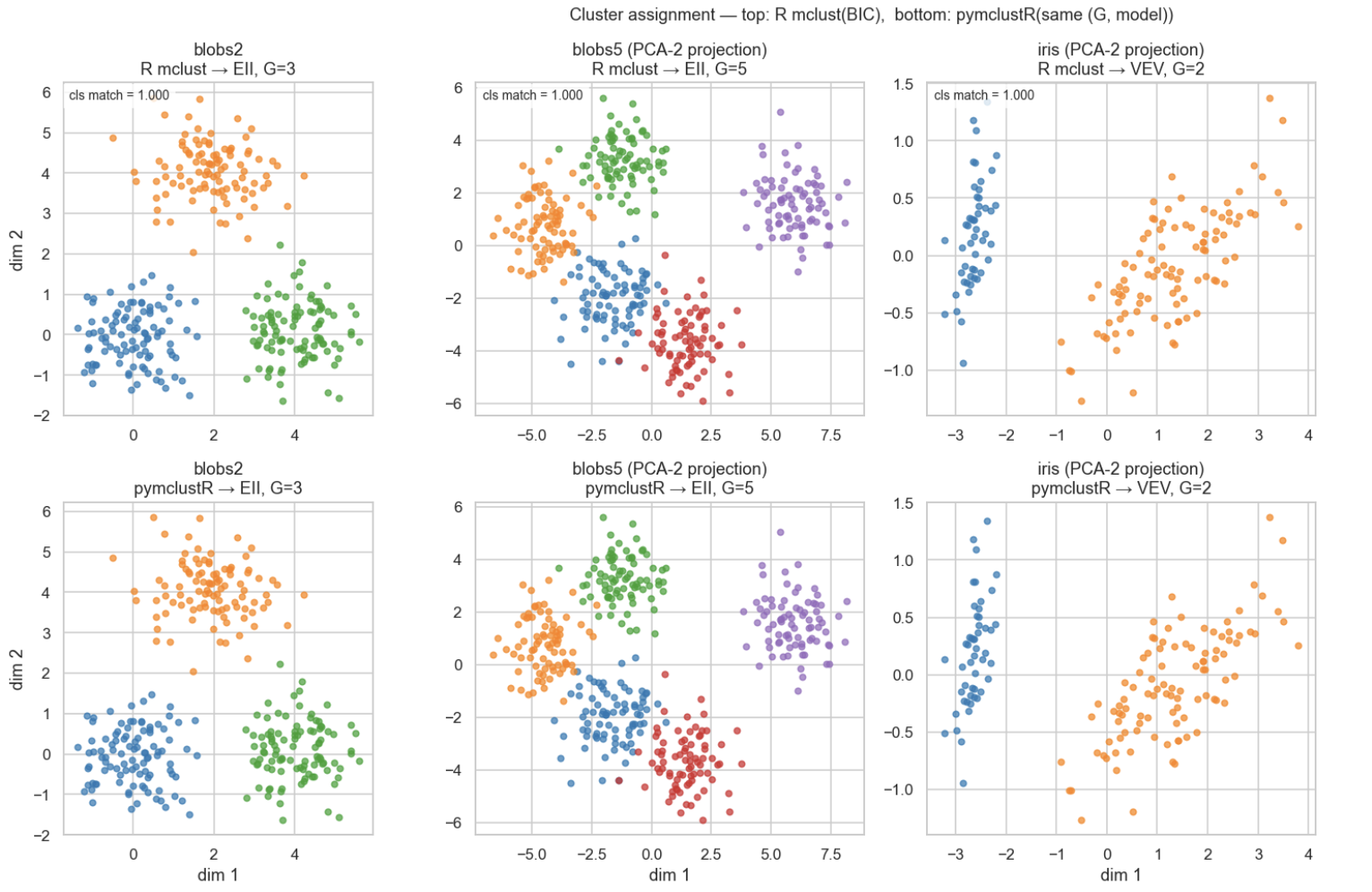
2.8 Added pymclustR to ov.utils.cluster, Fully Replacing mclust_R
I also like this update a lot.
In the new version, ov.utils.cluster adds method='pymclustR'. It is a pure Python reimplementation of mclust, replacing the previous mclust_R that depended on rpy2.
In the current version, the original mclust_R has been removed. Calling it will directly prompt users to use pymclustR.
This step represents a very clear recent direction for OmicVerse:
Wherever R dependencies can be removed, we should remove them as much as possible.
As these core algorithms are gradually Pythonized, the installation cost, calling cost, and tutorial barrier of the whole ecosystem will all decrease together.
Repository: https://github.com/omicverse/py-mclustR
2.9 Added the ov.metabol Metabolomics Module
This is a very important new boundary in 2.1.x.
We added ov.metabol. It is not just an entry point; it already begins to cover a relatively complete metabolomics analysis workflow:
- ID mapping
- lipidomics class summary
- MSEA
- Mummichog
- SERRF
- DGCA
- ASCA / MixedLM
- ROC / biomarker panel
This means OmicVerse's "multi-omics" in 2.1.x is no longer just a concept. It is truly starting to land in metabolomics.
Tutorial: https://omicverse.readthedocs.io/en/latest/tutorials/index_metabol.html
2.10 Added ov.micro and ov.alignment, Supporting a Full 16S / Microbiome Workflow
Another new boundary is microbiome analysis.
In this new version, we added ov.micro and ov.alignment, supporting a workflow from:
- cutadapt
- vsearch UNOISE3
- SINTAX
- MAFFT
- FastTree
all the way to:
- alpha / beta diversity
- differential abundance
- ordination
- taxonomy-level summary
In other words, this is no longer "supporting a few 16S functions." It is moving toward a complete microbiome workflow.
Tutorial: https://omicverse.readthedocs.io/en/latest/tutorials/index_microbiome.html
Postscript
Overall, this OmicVerse update means we are one step closer to a unified multi-omics ecosystem. For the development of a general bioinformatics Agent, it also means moving toward the next stage.
However, there are still many shortcomings and many areas worth improving. That is why the idea of building a community emerged. One person's power is limited after all. If you are interested in joining us, you can add my contact FernandoZeng and write this note: Name - Join Developer.
If you only want to provide ideas or suggestions for OmicVerse, you can also scan the group QR code below.
